End your hormonal struggles by treating the root causes with Functional Medicine
Reverse or manage endometriosis, PCOS, problem periods, fibroids, adenomyosis, PMDD, peri-menopause, menopause, vaginal infections and get help coming off the pill, with a Functional Medicine approach
Hormone imbalances, PCOS, endometriosis, fertility challenges, uterine fibroids, even breast and endometrial cancer, are more than just bad luck and faulty genes – they are the result of living in a chaotic environment.
Want to feel better?
You’ll have to go beyond the Pill, the coil, anti-depressants and surgery
You need to know why your hormones are unbalanced
If you go to the doctor’s office with menstrual pain, you get the Pill. If you go with a problematic cycle, you get the Pill. If you’re peri-menopausal you get anti-depressants.
Nobody is asking why do you have pain? Why do you have irregular cycles or heavy bleeding? Why do you have hot flushes and brain fog? What are the underlying factors?
The one-size-fits-all medical model is outdated
Treatments for women’s gynaecological health are still relegated to the 1950s: prescriptions for hormones, anti-depressants and surgery.
For some women these can be essential, but for the majority of women this approach can make things worse.
You are sensitive to food, toxins and stress
Research is clear: chronic use of hormonal contraceptives, chronic stress, antibiotics, steroids, infections, toxic overload (plastic, make up, junk food, sugar), nutritional deficiencies – these are the underlying causes of unbalanced hormones which need to be addressed to bring your body back into balance.
Hormones are messengers, and here’s what they’re saying
You’re undernourished and overfed
Your body needs a huge variety of vitamins, minerals and phytonutrients to function at it’s best.
What you do – or don’t eat – influences your menstrual cycle regularity, whether you experience PMS, menstrual pain, endometriosis, PCOS, fertility, uterine fibroids, breast health, and stressful moods. It shapes your long-term health, too – when you go into menopause, your bone strength, heart and brain health, and how you age.
Your metabolism is overwhelmed with toxins
Most of us have a moderate to even high body burden of any number of toxins from food, cosmetics, cleaning products and pollutions at or above levels known to cause adverse health effects.
Many of these are endocrine-disrupting chemicals, also called hormone disruptors, that interfere with the body’s endocrine system and overstimulate, block, or disrupt our hormone’s natural actions by sending mixed messages throughout your endocrine system.
You’re constantly stressed, inside and out
Is your stress response stuck on the On position?
When your stress response is constant on alert, it puts the brakes on ovarian and thyroid function. Even relatively short stretches of stress can impact your sex hormones and cycles. And the latest research on stress shows powerful links to irregular periods, menstrual pain, PMS, endometriosis, fertility challenges, PCOS, and more.
Functional Medicine is a breakthrough model for that connects your hormones to genes, food and lifestyle, and gives you the results you want
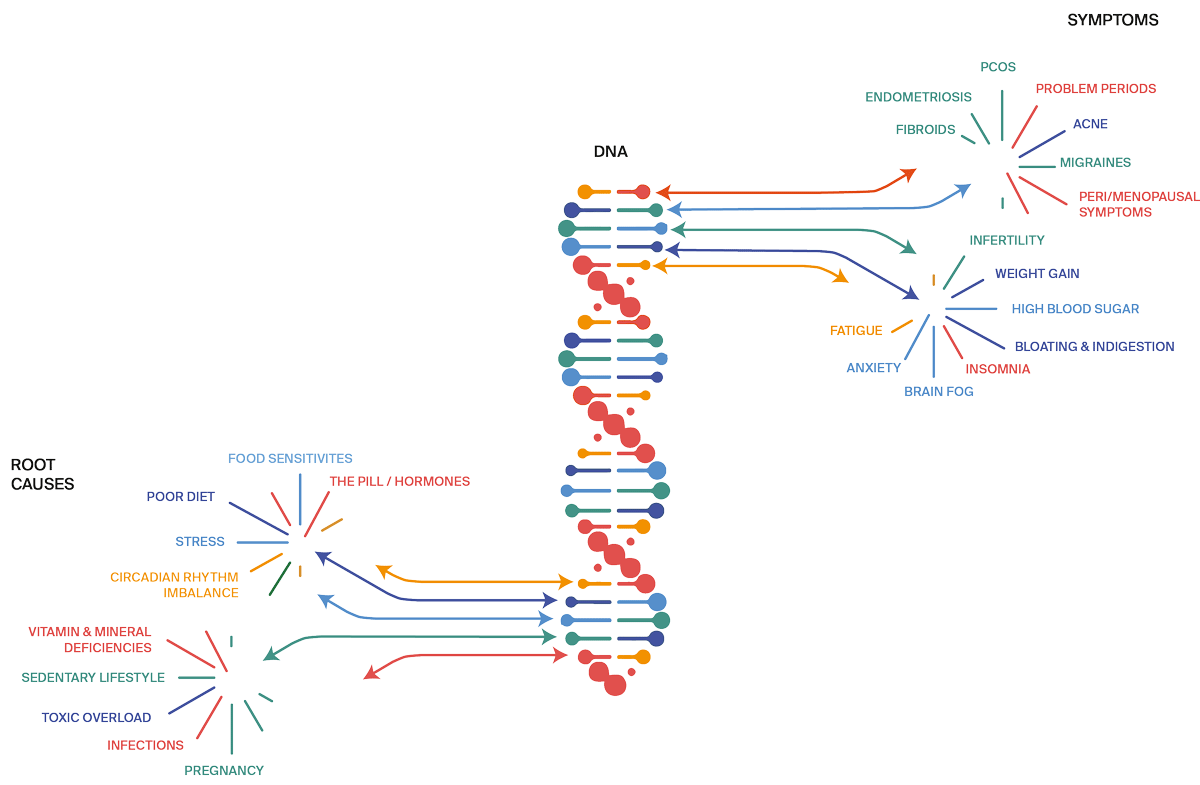
All your symptoms make perfect sense when understood in the context of your nutrition, genes, lifestyle, toxin and stress exposure.
Rooting out and treating the underlying causes of symptoms is key to getting better
This works because your hormonal system is logical and responsive to what you eat and how you live.
What do you need to get your health back on track?
Personalised nutrition and targeted supplements
Food is not just calories or energy; food is information, instructions that upgrade or downgrade your biology with every bite. It’s like code that programs your software. Your hardware is your genes. Your software is how those genes are turned on or off. Food regulates not only your genes but also your hormones, like insulin, testosterone, oestrogen, and thyroid. It alters your brain chemistry, producing happy chemicals, and it can even trigger addictive patterns.
You need a nourishing diet, but it needs to be personalised to you, to contain the nutrients, plant based chemicals, and ingredients that you need to support your brain, ovaries, uterus, adrenals, thyroid, while also supporting your gut, detoxification system, sleep, mind, and mood -supporting the inner ecosystems that create hormone balance.
Personalising your nutritional needs is key to optimising your health.
Detoxification
Almost all of us are experiencing a chronic combination of toxin overload and impaired detoxification, which is associated with low phytonutrient and phytochemical intakes.
Detoxification is a highly nutrient- and phytochemical-dependent process. It’s heavily reliant vitamins, minerals, and phytochemicals, so we have to keep them replenished to meet the demand. These include the B vitamins, choline, magnesium, selenium, zinc, folate, iron, calcium, copper, glutathione, sulphur, amino acids, and others. Insufficient amounts of any of these can inhibit the detoxification of not just numerous toxins, but also hormones like oestrogen. Oestrogen overload is the root cause of many conditions including PMS, fibroids, endometriosis and adenomyosis
Lightening your toxic exposures and supporting your natural detoxification pathways is the cornerstone of healing your hormonal imbalances.
A focus on self-care
Stress is almost synonymous with our modern lifestyles. We experience external stressors from our jobs, family responsibilities, financial worries, and many other places, then compound them with unhealthy lifestyle choices. The result is an epidemic of stress and increased risk for chronic hormonal conditions.
A focus on self-care and getting help with implementing stress relief techniques is the yin to the nutritional yang, speeding recovery and moving you towards health and joy.
Hormonal conditions respond particularly well to functional medicine and I can help
-357px.jpg)
Sandra Ishkanes
Women’s health expert
BSc MA DipION
I have a degree in molecular biology, clinical training in functional medicine and I am an expert in resolving the root causes of women’s hormonal conditions. I work like a health detective, where symptoms are my clues and poor health is my crime scene. I identify the root causes of unbalanced hormones and create solutions to optimise health.
Over 10 years of clinical experience
Innovation means a success rate of over 90%
Partnership, compassion and care
You want to get better, and I want to help you
1
Book a discovery call
Have a chat with me so you can decide if this approach is right for you and what it would look like to work together.
I usually make an initial assessment of where the root causes of your hormonal issues lie.
2
Follow your protocol
Have a consultation with to uncover the root causes of your hormonal imbalances.
Together we’ll create a plan that includes nutrition, supplements and lifestyle adjustments that work for you.
3
Results within 4 weeks
Succeed in alleviating pain, mood swings, balance your hormones and living life stress-free. Get help with coming off medication.
Start seeing results in as little as 4 weeks.
Case Studies
Wouldn’t it be amazing if there were was a logical approach to our health, rather than band-aids of anti-depressants, hormonal drugs and surgery? There is. Personalised nutrition, supplements and a focus on self-care effectively can naturally balance our hormones. And it’s scientifically proven.
-

Brain fog in menopause completely cleared in 8 weeks
LM was 59 years old when she came to see me asking for help with severe menopausal symptoms. Her most significant concerns were severe…
-

Lifelong depression and severe premenstrual anxiety
OT was 33 years when she got in touch with me seeking help for her long-standing daily anxiety and depression, which became much worse…
-

Acne and severe PMS
BA was 33 years old and working as a solicitor when she came to see me saying: She said: “Since the age of 13…
-

Premenstrual Dysphoric Disorder reversed in 8 weeks
TG was 43 when she came to see me about what she described as severe premenstrual syndrome (PMS): She said: “Two weeks before my…
My functional medicine approach is for you, if you have:
- PCOS, endometriosis, fibroids, adenomyosis
- Pelvic pain or breast pain
- Heavy periods
- Short cycles, long cycles or no periods
- Pre- or post-menstrual anxiety
- Difficulty managing your period, mood, pain and cyclical headaches/migraines and acne
- Recurrent vaginal or urinary infections
- Problems optimising your fertility
- A desire to optimise your health in peri-menopause and menopause – alleviate flooding, anxiety, brain fog, hot flushes, night sweats, joint pain, insomnia, rashes and vaginal dryness.